
最新刊期
卷 51 , 期 2 , 2026
-
 摘要:Electrocatalytic CO2 reduction reaction (CO2RR) provides an effective approach for converting CO2 into high value-added chemicals and fuels. In CO2RR research, precise modulation of the catalyst’s composition and structure is crucial for achieving high product selectivity. Cu is currently the most effective active metal component for driving CO2RR toward C2+ products. To gain a comprehensive understanding of the factors governing C2+ product formation on Cu-based catalysts, studies about CO2RR to C2+ products have been comprehensively analyzed and summarized. Starting from the adsorption mechanisms of CO2 on Cu-based catalyst surfaces, the effects of critical parameters such as Cu crystal facets, low-nuclearity Cu atomic clusters, Cu species valence states, and catalyst morphologies on CO2RR performance were thoroughly explored, with emphasis on their roles in regulating C2+ products selectivity. Furthermore, current challenges and future research directions for C2+ products by CO2RR were outlined, aiming to provide guidance for the rational design of high-efficiency catalysts for CO2RR.关键词:CO2RR;Cu-based catalysts;product selectivity;C2+ products47|0|0更新时间:2026-02-13
摘要:Electrocatalytic CO2 reduction reaction (CO2RR) provides an effective approach for converting CO2 into high value-added chemicals and fuels. In CO2RR research, precise modulation of the catalyst’s composition and structure is crucial for achieving high product selectivity. Cu is currently the most effective active metal component for driving CO2RR toward C2+ products. To gain a comprehensive understanding of the factors governing C2+ product formation on Cu-based catalysts, studies about CO2RR to C2+ products have been comprehensively analyzed and summarized. Starting from the adsorption mechanisms of CO2 on Cu-based catalyst surfaces, the effects of critical parameters such as Cu crystal facets, low-nuclearity Cu atomic clusters, Cu species valence states, and catalyst morphologies on CO2RR performance were thoroughly explored, with emphasis on their roles in regulating C2+ products selectivity. Furthermore, current challenges and future research directions for C2+ products by CO2RR were outlined, aiming to provide guidance for the rational design of high-efficiency catalysts for CO2RR.关键词:CO2RR;Cu-based catalysts;product selectivity;C2+ products47|0|0更新时间:2026-02-13 -
Study on preparation and Fischer-Tropsch synthesis performances of Mn or K modified
ε -Fe2C catalysts 摘要:Iron-based catalysts play a crucial role in Fischer-Tropsch Synthesis (FTS). However, their complex phase structures and high by-product selectivities limit further development. It is urgent to establish single-phase iron catalysts, explore the reaction processes and increase target product selectivities with promoters. Highly stable ε-Fe2C catalysts were successfully prepared by pyrolysis of Prussian Blue Analogues, and the effects of Mn and K promoters on the catalyst structures and catalytic performances were investigated. The results show that under reaction conditions of 270 °C, 3 MPa, n(H2):n(CO) = 2:1, and gas hourly space velocity (GHSV) of 30000 mL/(g·h), with the Mn-modified ε-Fe2C catalyst, CO2 selectivity decreases to 7.5%, and with the K-modified ε-Fe2C catalyst, C5+ hydrocarbon selectivity reaches 71.5% after 25 h reaction. The catalysts modified by the two promoters maintain ε-Fe2C structure before and after the reaction, and their activities and product selectivities remain stable during 80 h reaction. The Mn-modified ε-Fe2C catalyst exhibits better catalytic performance due to low by-product selectivity. By characterization methods such as XRD, TEM and XPS, the microstructural changes and their effects on FTS catalytic performances of the catalysts were revealed. It is found that the size effect produced by promoters results in a decrease in CO conversion rate with catalysts. Mn and K, as electronic promoters, provide electrons to the catalysts, ensuring the stability of the surface structures and regulating product distributions. The introduction of promoters into ε-Fe2C catalysts can effectively regulate catalytic activities and product distributions, providing a novel strategy for the development of FTS catalysts with high product selectivities.关键词:Fischer-Tropsch synthesis;iron carbide;promoter regulation;olefins66|0|0更新时间:2026-02-13
摘要:Iron-based catalysts play a crucial role in Fischer-Tropsch Synthesis (FTS). However, their complex phase structures and high by-product selectivities limit further development. It is urgent to establish single-phase iron catalysts, explore the reaction processes and increase target product selectivities with promoters. Highly stable ε-Fe2C catalysts were successfully prepared by pyrolysis of Prussian Blue Analogues, and the effects of Mn and K promoters on the catalyst structures and catalytic performances were investigated. The results show that under reaction conditions of 270 °C, 3 MPa, n(H2):n(CO) = 2:1, and gas hourly space velocity (GHSV) of 30000 mL/(g·h), with the Mn-modified ε-Fe2C catalyst, CO2 selectivity decreases to 7.5%, and with the K-modified ε-Fe2C catalyst, C5+ hydrocarbon selectivity reaches 71.5% after 25 h reaction. The catalysts modified by the two promoters maintain ε-Fe2C structure before and after the reaction, and their activities and product selectivities remain stable during 80 h reaction. The Mn-modified ε-Fe2C catalyst exhibits better catalytic performance due to low by-product selectivity. By characterization methods such as XRD, TEM and XPS, the microstructural changes and their effects on FTS catalytic performances of the catalysts were revealed. It is found that the size effect produced by promoters results in a decrease in CO conversion rate with catalysts. Mn and K, as electronic promoters, provide electrons to the catalysts, ensuring the stability of the surface structures and regulating product distributions. The introduction of promoters into ε-Fe2C catalysts can effectively regulate catalytic activities and product distributions, providing a novel strategy for the development of FTS catalysts with high product selectivities.关键词:Fischer-Tropsch synthesis;iron carbide;promoter regulation;olefins66|0|0更新时间:2026-02-13 -
 摘要:Traditional photocatalytic CO2 reduction systems typically require a large amount of substantial sacrificial agents to facilitate the reduction process. How to design efficient and stable catalysts to achieve a photocatalytic reaction without sacrificial agents has always been a research hotspot in this field. A series of novel Schottky heterojunction photocatalysts Ti3C2/Ag3VO4 (TAx, x = m(Ag3VO4)/m(Ti3C2)) were prepared by physical mixing method. Their structural properties were characterized by XRD, SEM and TEM, etc., and their photocatalytic CO2 reduction performance and mechanism were systematically studied. The results show that Ag3VO4 nanoparticles are uniformly dispersed onto the surface of Ti3C2 nanosheets, forming an intimate interfacial structure. Under sacrificial-agent-free conditions and visible light irradiation (λ ≥ 420 nm) for 3 h, TA1.5 exhibits the best photocatalytic CO2 reduction performance. The yields of CO and CH4 reach 80.67 μmol/g and 33.67 μmol/g, respectively. The yield of CO of TA1.5 is approximately 18.5 times and 17.2 times higher than that of Ti3C2 and Ag3VO4, respectively. The Schottky barrier formed between Ti3C2 and Ag3VO4 effectively promotes the directional migration of photogenerated electrons from Ag3VO4 to Ti3C2, thereby suppressing charge carrier recombination and significantly enhancing the photocatalytic CO2 reduction performance.关键词:Ti3C2 MXene;Ag3VO4;physical mixing;Schottky heterojunction;photocatalytic CO2 reduction0|0|0更新时间:2026-02-13
摘要:Traditional photocatalytic CO2 reduction systems typically require a large amount of substantial sacrificial agents to facilitate the reduction process. How to design efficient and stable catalysts to achieve a photocatalytic reaction without sacrificial agents has always been a research hotspot in this field. A series of novel Schottky heterojunction photocatalysts Ti3C2/Ag3VO4 (TAx, x = m(Ag3VO4)/m(Ti3C2)) were prepared by physical mixing method. Their structural properties were characterized by XRD, SEM and TEM, etc., and their photocatalytic CO2 reduction performance and mechanism were systematically studied. The results show that Ag3VO4 nanoparticles are uniformly dispersed onto the surface of Ti3C2 nanosheets, forming an intimate interfacial structure. Under sacrificial-agent-free conditions and visible light irradiation (λ ≥ 420 nm) for 3 h, TA1.5 exhibits the best photocatalytic CO2 reduction performance. The yields of CO and CH4 reach 80.67 μmol/g and 33.67 μmol/g, respectively. The yield of CO of TA1.5 is approximately 18.5 times and 17.2 times higher than that of Ti3C2 and Ag3VO4, respectively. The Schottky barrier formed between Ti3C2 and Ag3VO4 effectively promotes the directional migration of photogenerated electrons from Ag3VO4 to Ti3C2, thereby suppressing charge carrier recombination and significantly enhancing the photocatalytic CO2 reduction performance.关键词:Ti3C2 MXene;Ag3VO4;physical mixing;Schottky heterojunction;photocatalytic CO2 reduction0|0|0更新时间:2026-02-13 -
 摘要:Fe-based catalysts, owing to the low cost and excellent catalytic performance, are widely used in CO2 hydrogenation to light olefins. Na can significantly increase the selectivity of light olefins, but the underlying mechanism remains unclear. The influence of Na as a promoter on the reaction pathways and product relative selectivities of CO2 hydrogenation to light olefins on the Fe(111) surface was studied by combining density functional theory (DFT) calculations and microkinetic modeling (MKM) analysis. DFT calculations show that Na doping modifies the electron density of the Fe(111) surface and significantly promotes CO2 adsorption and activation. Moreover, Na reduces the energy barriers of C2H4* formation and desorption from 0.81 eV and 1.10 eV to 0.61 eV and 0.46 eV, respectively, and increase the energy barrier of CH4 formation from 0.95 eV to 1.15 eV. MKM analysis shows that Na doping substantially increases the rates of C2H4* formation and desorption and reduces the rate of CH4 formation. The rate-determining step of the entire reaction network shifts from C2H4* formation and desorption to CH4 formation. The above results reveal the intrinsic mechanism that Na increases the rates of C2H4 formation and reduces the rates of byproduct CH4 formation to increase the C2H4 relative selectivity by regulating electronic structures of the Fe(111) surface at the molecular level.关键词:CO2 hydrogenation;Fe-based catalysts;Na promoter;density functional theory;microkinetic modeling86|0|0更新时间:2026-02-13
摘要:Fe-based catalysts, owing to the low cost and excellent catalytic performance, are widely used in CO2 hydrogenation to light olefins. Na can significantly increase the selectivity of light olefins, but the underlying mechanism remains unclear. The influence of Na as a promoter on the reaction pathways and product relative selectivities of CO2 hydrogenation to light olefins on the Fe(111) surface was studied by combining density functional theory (DFT) calculations and microkinetic modeling (MKM) analysis. DFT calculations show that Na doping modifies the electron density of the Fe(111) surface and significantly promotes CO2 adsorption and activation. Moreover, Na reduces the energy barriers of C2H4* formation and desorption from 0.81 eV and 1.10 eV to 0.61 eV and 0.46 eV, respectively, and increase the energy barrier of CH4 formation from 0.95 eV to 1.15 eV. MKM analysis shows that Na doping substantially increases the rates of C2H4* formation and desorption and reduces the rate of CH4 formation. The rate-determining step of the entire reaction network shifts from C2H4* formation and desorption to CH4 formation. The above results reveal the intrinsic mechanism that Na increases the rates of C2H4 formation and reduces the rates of byproduct CH4 formation to increase the C2H4 relative selectivity by regulating electronic structures of the Fe(111) surface at the molecular level.关键词:CO2 hydrogenation;Fe-based catalysts;Na promoter;density functional theory;microkinetic modeling86|0|0更新时间:2026-02-13 -
 摘要:With the rapid growth of ethanol production, converting it into oxygenated chemicals and olefins can not only reduce dependence on petroleum resources, but also help meet the growing demand for energy and chemical products. Catalysts are the key to achieving efficient conversion of ethanol into oxygenated chemicals and olefins. Research progress on the reaction mechanisms, catalyst design strategies and catalytic performances of ethanol catalytic conversion to oxygenated chemicals and olefins was reviewed. And the future design of ethanol catalytic conversion catalysts was also prospected.关键词:ethanol;ethanol catalytic conversion catalysts;oxygenated chemicals;olefins278|0|0更新时间:2026-02-13
摘要:With the rapid growth of ethanol production, converting it into oxygenated chemicals and olefins can not only reduce dependence on petroleum resources, but also help meet the growing demand for energy and chemical products. Catalysts are the key to achieving efficient conversion of ethanol into oxygenated chemicals and olefins. Research progress on the reaction mechanisms, catalyst design strategies and catalytic performances of ethanol catalytic conversion to oxygenated chemicals and olefins was reviewed. And the future design of ethanol catalytic conversion catalysts was also prospected.关键词:ethanol;ethanol catalytic conversion catalysts;oxygenated chemicals;olefins278|0|0更新时间:2026-02-13 -
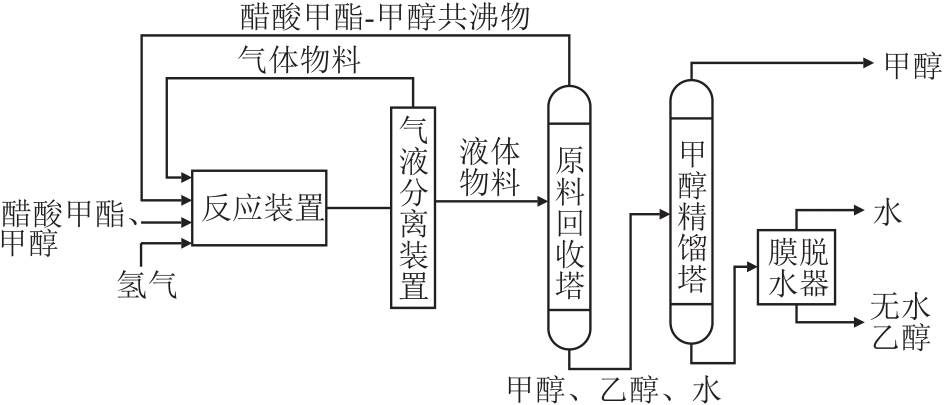 摘要:Methyl acetate, as an oxygen-containing organic solvent and chemical intermediate with high reactivity, has become an important pathway for improving carbon resource conversion efficiency and enhancing the added value of the chemical industry chain through its high-value utilization. Firstly, the preparation methods of methyl acetate and their principles and characteristics were reviewed, including processes such as methanol-acetic acid esterification, methanol carbonylation synthesis and methanol dehydrogenation coupling, and the energy consumption, costs and industrial applicability of different processes were compared. Then, focusing on the high-value utilization of methyl acetate, the main utilization routes and related technological progress were summarized, with particular emphasis on its development prospects in emerging application fields, which can provide new ideas for the utilization of methyl acetate. Finally, the existing problems in current high-value utilization technologies of methyl acetate were summarized, corresponding solutions were discussed, and suggestions for future research were proposed.关键词:methyl acetate;high-value utilization;Methyl Methacrylate;biodiesel20|0|0更新时间:2026-02-13
摘要:Methyl acetate, as an oxygen-containing organic solvent and chemical intermediate with high reactivity, has become an important pathway for improving carbon resource conversion efficiency and enhancing the added value of the chemical industry chain through its high-value utilization. Firstly, the preparation methods of methyl acetate and their principles and characteristics were reviewed, including processes such as methanol-acetic acid esterification, methanol carbonylation synthesis and methanol dehydrogenation coupling, and the energy consumption, costs and industrial applicability of different processes were compared. Then, focusing on the high-value utilization of methyl acetate, the main utilization routes and related technological progress were summarized, with particular emphasis on its development prospects in emerging application fields, which can provide new ideas for the utilization of methyl acetate. Finally, the existing problems in current high-value utilization technologies of methyl acetate were summarized, corresponding solutions were discussed, and suggestions for future research were proposed.关键词:methyl acetate;high-value utilization;Methyl Methacrylate;biodiesel20|0|0更新时间:2026-02-13 -
 摘要:Methanol-to-hydrocarbons (MTH) reaction serves as a “bridge” connecting coal chemical industry with petrochemical and natural gas chemical industries. HZSM-5 zeolite exhibits excellent catalytic performance in MTH reaction due to its unique pore structure and moderate to strong acidity. Formaldehyde can accelerate the MTH reaction process via the Prins reaction and also promote the cross-linking and condensation of poly-methyl benzenes to form polyaromatic hydrocarbons (PAH). However, the mechanism has not been thoroughly investigated. By designing a co-feeding experiment of toluene, benzene with formaldehyde on HZSM-5 zeolite, respectively, with combining gas chromatography-mass spectrometry and TGA, it is found that the key intermediate in the co-transformation of toluene and formaldehyde is diphenylmethane, which reveals the key interaction pathway between formaldehyde and toluene: Toluene and formaldehyde react at acid sites to form benzyl alcohol derivatives, which then dehydrate to form diphenylmethane. Diphenylmethane can not only accelerate the toluene conversion process but also act as a “bridge molecule” to significantly promote the cross-linking reaction of toluene.关键词:zeolite catalysis;toluene conversion;formaldehyde;coke-induced deactivation883|0|0更新时间:2026-02-13
摘要:Methanol-to-hydrocarbons (MTH) reaction serves as a “bridge” connecting coal chemical industry with petrochemical and natural gas chemical industries. HZSM-5 zeolite exhibits excellent catalytic performance in MTH reaction due to its unique pore structure and moderate to strong acidity. Formaldehyde can accelerate the MTH reaction process via the Prins reaction and also promote the cross-linking and condensation of poly-methyl benzenes to form polyaromatic hydrocarbons (PAH). However, the mechanism has not been thoroughly investigated. By designing a co-feeding experiment of toluene, benzene with formaldehyde on HZSM-5 zeolite, respectively, with combining gas chromatography-mass spectrometry and TGA, it is found that the key intermediate in the co-transformation of toluene and formaldehyde is diphenylmethane, which reveals the key interaction pathway between formaldehyde and toluene: Toluene and formaldehyde react at acid sites to form benzyl alcohol derivatives, which then dehydrate to form diphenylmethane. Diphenylmethane can not only accelerate the toluene conversion process but also act as a “bridge molecule” to significantly promote the cross-linking reaction of toluene.关键词:zeolite catalysis;toluene conversion;formaldehyde;coke-induced deactivation883|0|0更新时间:2026-02-13 -
 摘要:Producing low-carbon alcohols (C2~C6 alcohols) from syngas have received extensive attention due to its excellent economic property. The Co-based catalyst is one of the main catalysts for producing low-carbon alcohols from syngas, but the Co-based catalyst currently has the problem of low selectivity for low-carbon alcohols. Using the co-precipitation method, Ce and Mn modified Co/AC catalysts (Ce-Co/AC and Mn-Co/AC with Co mass fraction of 15.0% and Ce or Mn mass fraction of 7.5%) were prepared, respectively. The structures of the catalysts were characterized by XRD, N2 adsorption/desorption and H2-TPR, and the catalytic performances of catalysts for producing low-carbon alcohols from syngas (calculating by the average values after 7 d of reaction) under the conditions of temperature of 250 ℃, pressure of 3.0 MPa and syngas (V(H2):V(CO) = 2:1) flow rate of 75 mL/min were evaluated. The results show that compared with Co/AC, the introduction of the additives Ce and Mn both can improve the dispersion of Co species on the catalyst surface and reduce the crystal size of Co0 species. Among them, the content (mole fraction) of surface Co0 species in Mn-Co/AC is 66.0%. Under the experimental conditions, the CO conversion rate of Ce-Co/AC is 2.29%, and the total alcohol selectivity is 32.03% and m(low-carbon alcohols)/m(total alcohols) is 68.06%. Mn-Co/AC exhibits better catalytic performance with CO conversion rate of 3.99%, total alcohol selectivity of 33.23% and m(low-carbon alcohols)/m(total alcohols) of 79.36%.关键词:Co-based catalyst;co-precipitation method;syngas;Co species;low carbon alcohols41|0|0更新时间:2026-02-13
摘要:Producing low-carbon alcohols (C2~C6 alcohols) from syngas have received extensive attention due to its excellent economic property. The Co-based catalyst is one of the main catalysts for producing low-carbon alcohols from syngas, but the Co-based catalyst currently has the problem of low selectivity for low-carbon alcohols. Using the co-precipitation method, Ce and Mn modified Co/AC catalysts (Ce-Co/AC and Mn-Co/AC with Co mass fraction of 15.0% and Ce or Mn mass fraction of 7.5%) were prepared, respectively. The structures of the catalysts were characterized by XRD, N2 adsorption/desorption and H2-TPR, and the catalytic performances of catalysts for producing low-carbon alcohols from syngas (calculating by the average values after 7 d of reaction) under the conditions of temperature of 250 ℃, pressure of 3.0 MPa and syngas (V(H2):V(CO) = 2:1) flow rate of 75 mL/min were evaluated. The results show that compared with Co/AC, the introduction of the additives Ce and Mn both can improve the dispersion of Co species on the catalyst surface and reduce the crystal size of Co0 species. Among them, the content (mole fraction) of surface Co0 species in Mn-Co/AC is 66.0%. Under the experimental conditions, the CO conversion rate of Ce-Co/AC is 2.29%, and the total alcohol selectivity is 32.03% and m(low-carbon alcohols)/m(total alcohols) is 68.06%. Mn-Co/AC exhibits better catalytic performance with CO conversion rate of 3.99%, total alcohol selectivity of 33.23% and m(low-carbon alcohols)/m(total alcohols) of 79.36%.关键词:Co-based catalyst;co-precipitation method;syngas;Co species;low carbon alcohols41|0|0更新时间:2026-02-13 -
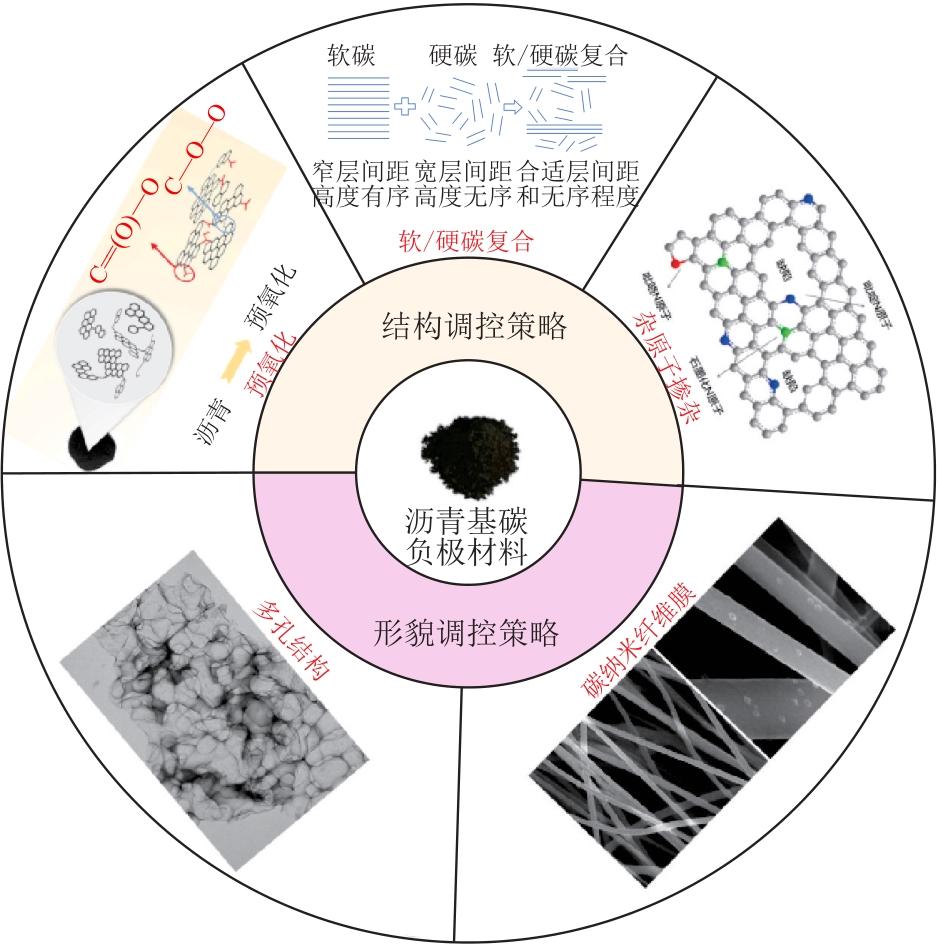 摘要:As a high-carbon material rich in aromatic structures, asphalt stands out as an excellent precursor material for sodium-ion battery carbon anodes due to its advantages of wide availability, high carbon content and low cost. However, asphalt-based carbon materials prepared by direct pyrolysis suffer from disadvantages such as high graphitization and narrow interlayer spacing, which limit sodium-ion storage capacity. Overcoming these limitations through structural and morphological regulations has become a key research focus in the field. Research progress on asphalt-based carbon anode materials were reviewed. The technical routes and modification mechanisms of structural regulation (pre-oxidation, soft/hard carbon composite and heteroatom doping) and morphological regulation (construction of porous structures and carbon nanofiber membranes) was mainly introduced. The structure-property relationship between microstructure and sodium storage performance and the advantages and disadvantages of various regulation methods were analyzed. Finally, insights into future research directions of asphalt-based carbon anode materials were provided from the perspectives of fundamental science, material design and industrial application, respectively.关键词:sodium-ion batteries;asphalt-based carbon anode materials;structural regulation;morphological regulation91|0|0更新时间:2026-02-13
摘要:As a high-carbon material rich in aromatic structures, asphalt stands out as an excellent precursor material for sodium-ion battery carbon anodes due to its advantages of wide availability, high carbon content and low cost. However, asphalt-based carbon materials prepared by direct pyrolysis suffer from disadvantages such as high graphitization and narrow interlayer spacing, which limit sodium-ion storage capacity. Overcoming these limitations through structural and morphological regulations has become a key research focus in the field. Research progress on asphalt-based carbon anode materials were reviewed. The technical routes and modification mechanisms of structural regulation (pre-oxidation, soft/hard carbon composite and heteroatom doping) and morphological regulation (construction of porous structures and carbon nanofiber membranes) was mainly introduced. The structure-property relationship between microstructure and sodium storage performance and the advantages and disadvantages of various regulation methods were analyzed. Finally, insights into future research directions of asphalt-based carbon anode materials were provided from the perspectives of fundamental science, material design and industrial application, respectively.关键词:sodium-ion batteries;asphalt-based carbon anode materials;structural regulation;morphological regulation91|0|0更新时间:2026-02-13 -
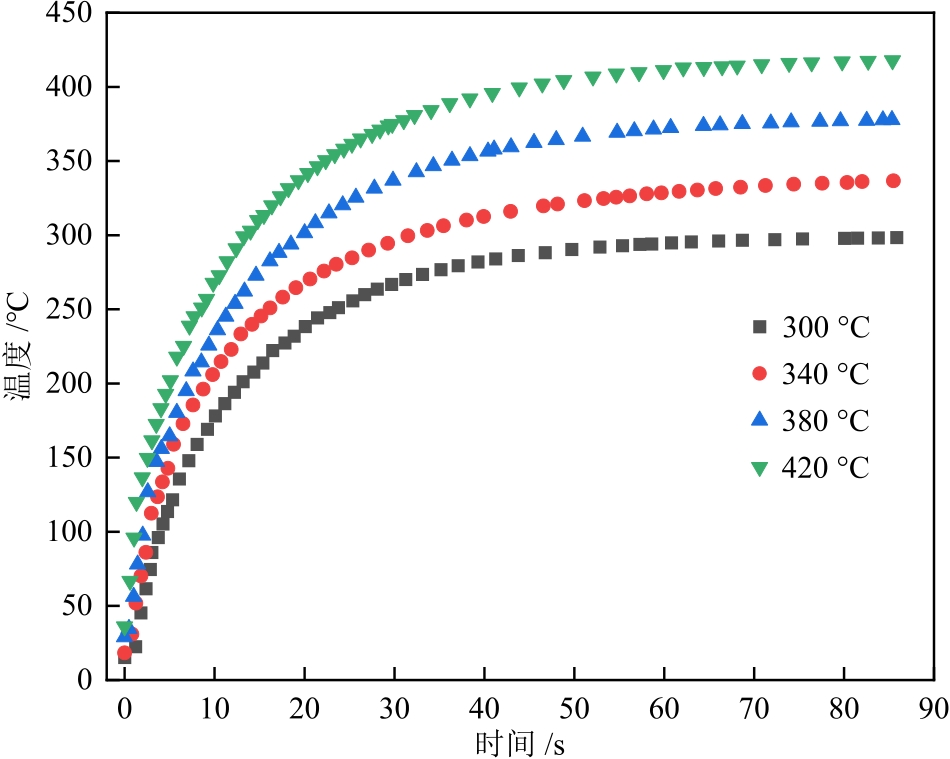 摘要:As pyrolysis serves as the important technology for the comprehensive utilization of corn stalks (hereinafter referred to as “stalks”), the process follows a free radical reaction mechanism. Understanding the compositions of covalent bonds in stalks and the generation characteristics of free radicals during pyrolysis is of great significance for elucidating the underlying mechanisms of free radical reactions and their relationships with organic structures, thereby contributing to the optimization of pyrolysis processes. The contents of major covalent bonds in stalks were calculated based on elemental analysis and carbon skeleton structural parameters derived from 13C NMR characterization. 9,10-dihydroanthracene was employed as a molecular probe to indirectly quantify the formation of active free radicals during pyrolysis. Additionally, the numbers and g-values of stable free radicals formed after pyrolysis were determined using electron spin resonance (ESR) spectroscopy. The results show that the contents of Cal—Cal and Car—Car in stalks are 28.47 mmol/g and 4.95 mmol/g, respectively, with Cal—O (with a content of up to 28.57 mmol/g) as the dominant C—O type. The concentration of active free radicals (cR-A) gradually increases with higher temperature and longer residence time, reaching 4.60 mmol/g to 29.44 mmol/g after 12 min of pyrolysis at 300 ℃ to 420 °C. During pyrolysis of stalks, weak bonds such as β-O—Cal and β-Cal—Cal are likely to be cleaved, generating active free radicals primarily consisting of aliphatic carbon radicals, oxygen radicals bonded to aromatic carbons, and aliphatic carbon radicals bonded to aromatic carbons. As the pyrolysis of stalks proceeds, the concentration of stable free radicals first increases rapidly, then decreases slowly, and finally increases slightly again. Large-sized active radicals of aromatic structures (especially ·Cal—Car) are more likely to transform into stable free radicals during coupling. The g-value of newly formed stable free radicals gradually decreases with the extension of pyrolysis time, with more oxygen-containing radicals.关键词:corn stalks;covalent bonds;pyrolysis;free radicals;g-values113|0|0更新时间:2026-02-13
摘要:As pyrolysis serves as the important technology for the comprehensive utilization of corn stalks (hereinafter referred to as “stalks”), the process follows a free radical reaction mechanism. Understanding the compositions of covalent bonds in stalks and the generation characteristics of free radicals during pyrolysis is of great significance for elucidating the underlying mechanisms of free radical reactions and their relationships with organic structures, thereby contributing to the optimization of pyrolysis processes. The contents of major covalent bonds in stalks were calculated based on elemental analysis and carbon skeleton structural parameters derived from 13C NMR characterization. 9,10-dihydroanthracene was employed as a molecular probe to indirectly quantify the formation of active free radicals during pyrolysis. Additionally, the numbers and g-values of stable free radicals formed after pyrolysis were determined using electron spin resonance (ESR) spectroscopy. The results show that the contents of Cal—Cal and Car—Car in stalks are 28.47 mmol/g and 4.95 mmol/g, respectively, with Cal—O (with a content of up to 28.57 mmol/g) as the dominant C—O type. The concentration of active free radicals (cR-A) gradually increases with higher temperature and longer residence time, reaching 4.60 mmol/g to 29.44 mmol/g after 12 min of pyrolysis at 300 ℃ to 420 °C. During pyrolysis of stalks, weak bonds such as β-O—Cal and β-Cal—Cal are likely to be cleaved, generating active free radicals primarily consisting of aliphatic carbon radicals, oxygen radicals bonded to aromatic carbons, and aliphatic carbon radicals bonded to aromatic carbons. As the pyrolysis of stalks proceeds, the concentration of stable free radicals first increases rapidly, then decreases slowly, and finally increases slightly again. Large-sized active radicals of aromatic structures (especially ·Cal—Car) are more likely to transform into stable free radicals during coupling. The g-value of newly formed stable free radicals gradually decreases with the extension of pyrolysis time, with more oxygen-containing radicals.关键词:corn stalks;covalent bonds;pyrolysis;free radicals;g-values113|0|0更新时间:2026-02-13 -
 摘要:Chloride impurities in hydrogen can cause irreversible damage to hydrogen fuel cells. To improve the performance of Ni/NaY adsorbents for removing dichloromethane from hydrogen at room temperature, a series of Fe-Ni/NaY adsorbents with different Fe loadings (mass fraction, the same below) were prepared by equal-volume impregnation method. The adsorption performance of the Fe-Ni/NaY adsorbents for dichloromethane in hydrogen was investigated using a fixed-bed reactor, and the adsorbents were characterized by XRD, FT-IR and NH3-TPD. The results show that the introduction of Fe promoter increases the specific surface area and micropore surface area of the Fe-Ni/NaY adsorbent, and increases the number of weak acid sites of the NaY molecular sieve. As the Fe loading increases, the number of weak acid sites increases significantly, thereby enhancing the adsorption capacity of the adsorbent for removing dichloromethane in hydrogen at room temperature. When the Fe loading is 0.15%, under the conditions of 25 ℃, 0.1 MPa and a hydrogen flow rate of 6000 mL/h, the chlorine adsorption capacity of the Fe-Ni/NaY adsorbent reaches 5.36%.关键词:hydrogen;dichloromethane;adsorption;Ni/NaY adsorbent;Fe promoter31|0|0更新时间:2026-02-13
摘要:Chloride impurities in hydrogen can cause irreversible damage to hydrogen fuel cells. To improve the performance of Ni/NaY adsorbents for removing dichloromethane from hydrogen at room temperature, a series of Fe-Ni/NaY adsorbents with different Fe loadings (mass fraction, the same below) were prepared by equal-volume impregnation method. The adsorption performance of the Fe-Ni/NaY adsorbents for dichloromethane in hydrogen was investigated using a fixed-bed reactor, and the adsorbents were characterized by XRD, FT-IR and NH3-TPD. The results show that the introduction of Fe promoter increases the specific surface area and micropore surface area of the Fe-Ni/NaY adsorbent, and increases the number of weak acid sites of the NaY molecular sieve. As the Fe loading increases, the number of weak acid sites increases significantly, thereby enhancing the adsorption capacity of the adsorbent for removing dichloromethane in hydrogen at room temperature. When the Fe loading is 0.15%, under the conditions of 25 ℃, 0.1 MPa and a hydrogen flow rate of 6000 mL/h, the chlorine adsorption capacity of the Fe-Ni/NaY adsorbent reaches 5.36%.关键词:hydrogen;dichloromethane;adsorption;Ni/NaY adsorbent;Fe promoter31|0|0更新时间:2026-02-13 -
 摘要:Coking wastewater, characterized by its complex composition, is a typical industrial wastewater with a high concentration of chemical oxygen demand (COD), which is resistant to biodegradation. Currently, most coking plants struggle to achieve compliance standards for COD even after wastewater treatment, thus there is an urgent need to develop a new type of green, efficient and low-cost adsorbent. A series of modified amorphous silica-alumina adsorbents were synthesized via the co-precipitation method and applied for COD removal in coking wastewater. By comparing the COD removal rates of the original adsorbent and modified adsorbents through drying, calcination, and ammonium exchange (ASA-1, ASA-2, ASA-3, and ASA-4), the influence mechanisms of different modification methods on the physicochemical properties and adsorption performances of the adsorbents were explored. The results show that ASA-3 exhibits the best COD removal efficiency among the four adsorbents. At a ratio of silicon atoms number to aluminium atoms number of 4.50, calcination temperature of 750 ℃, and dosage of 50 g/L, COD removal reaches 62.40% after stirring for 3 h at 25 ℃. The excellent adsorption performance of ASA-3 is attributed to its enhanced textural properties (specific surface area of 271.0 m2/g) and abundant acidic sites. Conversely, although the acidic sites of ASA-4 increase, its adsorption performance is relatively poor (with a COD removal rate of only 32.54%). This is because the ammonium exchange process disrupt its structure (a specific surface area of merely 53.8 m2/g). Adsorption capacities of modified adsorbents at different adsorption time were fitted using pseudo-first-order, pseudo-second-order, and intra-particle diffusion models. The adsorption mechanism was investigated, revealing that the adsorption process conforms to the pseudo-second-order model and is governed by liquid film diffusion, chemical adsorption, and intra-particle diffusion mechanisms.关键词:amorphous silicon-aluminum;adsorption;coking wastewater;COD;kinetics0|0|0更新时间:2026-02-13
摘要:Coking wastewater, characterized by its complex composition, is a typical industrial wastewater with a high concentration of chemical oxygen demand (COD), which is resistant to biodegradation. Currently, most coking plants struggle to achieve compliance standards for COD even after wastewater treatment, thus there is an urgent need to develop a new type of green, efficient and low-cost adsorbent. A series of modified amorphous silica-alumina adsorbents were synthesized via the co-precipitation method and applied for COD removal in coking wastewater. By comparing the COD removal rates of the original adsorbent and modified adsorbents through drying, calcination, and ammonium exchange (ASA-1, ASA-2, ASA-3, and ASA-4), the influence mechanisms of different modification methods on the physicochemical properties and adsorption performances of the adsorbents were explored. The results show that ASA-3 exhibits the best COD removal efficiency among the four adsorbents. At a ratio of silicon atoms number to aluminium atoms number of 4.50, calcination temperature of 750 ℃, and dosage of 50 g/L, COD removal reaches 62.40% after stirring for 3 h at 25 ℃. The excellent adsorption performance of ASA-3 is attributed to its enhanced textural properties (specific surface area of 271.0 m2/g) and abundant acidic sites. Conversely, although the acidic sites of ASA-4 increase, its adsorption performance is relatively poor (with a COD removal rate of only 32.54%). This is because the ammonium exchange process disrupt its structure (a specific surface area of merely 53.8 m2/g). Adsorption capacities of modified adsorbents at different adsorption time were fitted using pseudo-first-order, pseudo-second-order, and intra-particle diffusion models. The adsorption mechanism was investigated, revealing that the adsorption process conforms to the pseudo-second-order model and is governed by liquid film diffusion, chemical adsorption, and intra-particle diffusion mechanisms.关键词:amorphous silicon-aluminum;adsorption;coking wastewater;COD;kinetics0|0|0更新时间:2026-02-13 -
Preparation of A/X-type and X-type molecular sieves from fly ash and their CO2 adsorption properties
 摘要:Resource utilization of industrial wastes is an important topic in the field of sustainable development, among which the high value-added utilization of fly ash is particularly important. Fly ash with different Si/Al ratios (n(Si)/n(Al) of 2.29 and 1.42, respectively) from two thermal power plants in Qingdao (Q) and Zibo (Z) was used as raw material. Molecular sieves for CO2 adsorption were synthesized by the alkali fusion-hydrothermal method. XRD, XRF and SEM were employed to characterize the composition and structural properties of fly ash and molecular sieves. In combination with one-factor experiments, the optimal treatment and synthesis conditions were determined, and the effects of alkali-to-ash ratio (mass ratio), liquid-to-solid ratio (mass ratio) and crystallization time on the synthesis of A/X-type and X-type molecular sieves were investigated. The results show that, due to the different Si/Al ratios of the fly ashes, the optimal treatment and synthesis conditions also differ. Under the optimal conditions of an alkali-to-ash ratio of 1.6:1, a liquid-to-solid ratio of 8:1 and a crystallization time of 6 h, the X-type molecular sieve synthesized from fly ash Q exhibits a specific surface area of 532.7 m2/g, with dynamic and static CO2 adsorption capacities of 2.77 mmol/g and 4.42 mmol/g, respectively. These values are significantly higher than those of the A/X-type molecular sieve synthesized from fly ash Z under its optimal conditions (281.2 m2/g, 1.57 mmol/g and 3.95 mmol/g, respectively). Adsorption kinetics studies indicate that the CO2 adsorption process of both molecular sieves is dominated by physical adsorption. The X-type molecular sieve maintains over 85% of its initial CO2 adsorption capacity after 10 adsorption/desorption cycles. The Si/Al ratio of fly ash is identified as the key factor determining the performance of the molecular sieves. These findings provide a reference for the preparation of high-performance molecular sieve adsorbents from fly ash.关键词:fly ash;A/X-type molecular sieve;X-type molecular sieve;CO2 adsorption35|0|0更新时间:2026-02-13
摘要:Resource utilization of industrial wastes is an important topic in the field of sustainable development, among which the high value-added utilization of fly ash is particularly important. Fly ash with different Si/Al ratios (n(Si)/n(Al) of 2.29 and 1.42, respectively) from two thermal power plants in Qingdao (Q) and Zibo (Z) was used as raw material. Molecular sieves for CO2 adsorption were synthesized by the alkali fusion-hydrothermal method. XRD, XRF and SEM were employed to characterize the composition and structural properties of fly ash and molecular sieves. In combination with one-factor experiments, the optimal treatment and synthesis conditions were determined, and the effects of alkali-to-ash ratio (mass ratio), liquid-to-solid ratio (mass ratio) and crystallization time on the synthesis of A/X-type and X-type molecular sieves were investigated. The results show that, due to the different Si/Al ratios of the fly ashes, the optimal treatment and synthesis conditions also differ. Under the optimal conditions of an alkali-to-ash ratio of 1.6:1, a liquid-to-solid ratio of 8:1 and a crystallization time of 6 h, the X-type molecular sieve synthesized from fly ash Q exhibits a specific surface area of 532.7 m2/g, with dynamic and static CO2 adsorption capacities of 2.77 mmol/g and 4.42 mmol/g, respectively. These values are significantly higher than those of the A/X-type molecular sieve synthesized from fly ash Z under its optimal conditions (281.2 m2/g, 1.57 mmol/g and 3.95 mmol/g, respectively). Adsorption kinetics studies indicate that the CO2 adsorption process of both molecular sieves is dominated by physical adsorption. The X-type molecular sieve maintains over 85% of its initial CO2 adsorption capacity after 10 adsorption/desorption cycles. The Si/Al ratio of fly ash is identified as the key factor determining the performance of the molecular sieves. These findings provide a reference for the preparation of high-performance molecular sieve adsorbents from fly ash.关键词:fly ash;A/X-type molecular sieve;X-type molecular sieve;CO2 adsorption35|0|0更新时间:2026-02-13 -
 摘要:In both the extraction and transportation of helium-rich natural gas (HNG) and the development of the new hydrate-based helium (He) separation technology, the gas-liquid systems involved in the fields always have the components that can form hydrate. The combination of the effects of those components on hydrate formation makes it hard to achieve the accurate prediction of the critical formation pressure (peq) of HNG hydrate. To solve the problem, the effects of the volume fractions of He, nitrogen (N2) and carbon dioxide (CO2) in gas phase, as well as the mass fraction of tetrahydrofuran (THF) in liquid phase on peq were investigated through the thermodynamic phase equilibrium experiments. Based on the experimental investigation, a new model which can predict the peq was proposed. The results show that the performance of THF on decreasing peq becomes more significant as the volume fraction of He increases. As the volume fraction of N2 continuously increases, the performance of THF on decreasing peq keeps constant at first then increases. As the volume fraction of CO2 continuously increases, the performance of THF on decreasing peq becomes slightly weak at first then keeps constant. The positive effect of CO2 on gas hydrate formation is stronger than that of methane (CH4), whereas the positive effect of CO2 on THF hydrate formation is weaker than that of CH4. The modeling results show good accuracy on predicting peq, with the average relative deviation between the modeling peq and experimental peq of 2.6%, and the fit goodness of of the critical hydrate formation temperature-pressure curve of each gas-liquid system is higher than 0.965.关键词:helium-rich natural gas;hydrates;prediction model;thermodynamic equilibrium;critical formation conditions14|0|0更新时间:2026-02-13
摘要:In both the extraction and transportation of helium-rich natural gas (HNG) and the development of the new hydrate-based helium (He) separation technology, the gas-liquid systems involved in the fields always have the components that can form hydrate. The combination of the effects of those components on hydrate formation makes it hard to achieve the accurate prediction of the critical formation pressure (peq) of HNG hydrate. To solve the problem, the effects of the volume fractions of He, nitrogen (N2) and carbon dioxide (CO2) in gas phase, as well as the mass fraction of tetrahydrofuran (THF) in liquid phase on peq were investigated through the thermodynamic phase equilibrium experiments. Based on the experimental investigation, a new model which can predict the peq was proposed. The results show that the performance of THF on decreasing peq becomes more significant as the volume fraction of He increases. As the volume fraction of N2 continuously increases, the performance of THF on decreasing peq keeps constant at first then increases. As the volume fraction of CO2 continuously increases, the performance of THF on decreasing peq becomes slightly weak at first then keeps constant. The positive effect of CO2 on gas hydrate formation is stronger than that of methane (CH4), whereas the positive effect of CO2 on THF hydrate formation is weaker than that of CH4. The modeling results show good accuracy on predicting peq, with the average relative deviation between the modeling peq and experimental peq of 2.6%, and the fit goodness of of the critical hydrate formation temperature-pressure curve of each gas-liquid system is higher than 0.965.关键词:helium-rich natural gas;hydrates;prediction model;thermodynamic equilibrium;critical formation conditions14|0|0更新时间:2026-02-13 -
 摘要:Kinetics enhancement of CO2 hydrate formation is the key to improving the efficiency of CO2 capture via the hydrate-based method. The kinetic characteristics of CO2 hydrate formation in 1,3-dioxolane (DIOX) solution were investigated from both macro- and microscopic perspectives, including the effects of gas-liquid ratio (ratio of gas-phase volume to liquid-phase volume in the reactor), reaction temperature and DIOX concentration (mole fraction, the same below) on key kinetic parameters such as induction time and gas consumption. The mechanism of CO2 hydrate formation in DIOX solution was also explored using a microscopic visualization system. The results show that increasing the gas-liquid ratio enhances CO2 mass transfer in the liquid phase and promotes CO2 hydrate formation, with the shortest induction time of 18 min obtained at a gas-liquid ratio of 2.33. Increasing the reaction temperature can increase gas consumption, but excessively high temperatures can reduce the hydrate formation rate. The maximum gas consumption of 26.04 mmol/mol is achieved at 277.15 K. Increasing the DIOX concentration promotes CO2 hydrate formation, with the best promotion effect observed at 5.56%. At the initial stage of the experiment, CO2 rapidly dissolves in the DIOX solution, and hydrates first form at the gas-liquid interface. The hydrates then grow continuously downward, forming pasty hydrates and exhibiting a spatial distribution characterized by a dense upper layer and a sparse lower layer. This study provides a theoretical basis for optimizing the kinetics of CO2 hydrate formation.关键词:CO2 hydrate;DIOX solution;kinetic characteristics;CO2 capture59|0|0更新时间:2026-02-13
摘要:Kinetics enhancement of CO2 hydrate formation is the key to improving the efficiency of CO2 capture via the hydrate-based method. The kinetic characteristics of CO2 hydrate formation in 1,3-dioxolane (DIOX) solution were investigated from both macro- and microscopic perspectives, including the effects of gas-liquid ratio (ratio of gas-phase volume to liquid-phase volume in the reactor), reaction temperature and DIOX concentration (mole fraction, the same below) on key kinetic parameters such as induction time and gas consumption. The mechanism of CO2 hydrate formation in DIOX solution was also explored using a microscopic visualization system. The results show that increasing the gas-liquid ratio enhances CO2 mass transfer in the liquid phase and promotes CO2 hydrate formation, with the shortest induction time of 18 min obtained at a gas-liquid ratio of 2.33. Increasing the reaction temperature can increase gas consumption, but excessively high temperatures can reduce the hydrate formation rate. The maximum gas consumption of 26.04 mmol/mol is achieved at 277.15 K. Increasing the DIOX concentration promotes CO2 hydrate formation, with the best promotion effect observed at 5.56%. At the initial stage of the experiment, CO2 rapidly dissolves in the DIOX solution, and hydrates first form at the gas-liquid interface. The hydrates then grow continuously downward, forming pasty hydrates and exhibiting a spatial distribution characterized by a dense upper layer and a sparse lower layer. This study provides a theoretical basis for optimizing the kinetics of CO2 hydrate formation.关键词:CO2 hydrate;DIOX solution;kinetic characteristics;CO2 capture59|0|0更新时间:2026-02-13

- Postal code:610225
- Tel:028-85964717 Email:236764603@qq.com
- Technical support is provided by Beijing Founder electronics co., LTD 蜀ICP备12008600号-10
 蜀公网安备51012202001533
蜀公网安备51012202001533 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
- Total Visits:378882 Visits Today:30

0