
最新刊期
卷 50 , 期 12 , 2025
-
 摘要:Electrocatalytic CO2 reduction reaction (ECO2RR) for producing high value-added chemicals and fuels is an effective approach to achieving “dual carbon” goals, with formic acid (HCOOH) being one of the most economically valuable products. However, the thermodynamic stability of CO2 molecules and the multi-proton-electron coupling processes during ECO2RR limit the reaction activity and product selectivity. p-block metal-based (such as Bi, In, Sn, etc.) catalysts show significant potential in ECO2RR to HCOOH, owing to their appropriate key formic acid intermediate OCHO* adsorption capability. Under electrochemical operating conditions, the catalyst surface undergoes reconstruction induced by electric fields, interfacial reactions and changes of chemical environments. This reconstruction makes it difficult to reveal the relationship between the structure and catalytic performance of catalysts. In light of this, focusing on the in situ reconstruction behavior of p-block metal-based catalysts during ECO2RR, the influencing factors, reconstruction types and characterization techniques were systematically analyzed, and the structure-performance relationships of catalysts under dynamic operating conditions were explored, and different reconstruction pathways and evolution laws of active sites of p-block metal-based catalysts during ECO2RR to HCOOH were summarized. A suggestion was put forward to construct a “theoretical calculation-machine learning-in situ experiment” system, providing a reference for designing reconstructed catalysts with high stability and product selectivity.关键词:electrocatalytic CO2 reduction;formic acid;p-block metal-based catalysts;in situ reconstruction of catalysts;reconstruction types;in situ characterization techniques94|0|0更新时间:2025-12-25
摘要:Electrocatalytic CO2 reduction reaction (ECO2RR) for producing high value-added chemicals and fuels is an effective approach to achieving “dual carbon” goals, with formic acid (HCOOH) being one of the most economically valuable products. However, the thermodynamic stability of CO2 molecules and the multi-proton-electron coupling processes during ECO2RR limit the reaction activity and product selectivity. p-block metal-based (such as Bi, In, Sn, etc.) catalysts show significant potential in ECO2RR to HCOOH, owing to their appropriate key formic acid intermediate OCHO* adsorption capability. Under electrochemical operating conditions, the catalyst surface undergoes reconstruction induced by electric fields, interfacial reactions and changes of chemical environments. This reconstruction makes it difficult to reveal the relationship between the structure and catalytic performance of catalysts. In light of this, focusing on the in situ reconstruction behavior of p-block metal-based catalysts during ECO2RR, the influencing factors, reconstruction types and characterization techniques were systematically analyzed, and the structure-performance relationships of catalysts under dynamic operating conditions were explored, and different reconstruction pathways and evolution laws of active sites of p-block metal-based catalysts during ECO2RR to HCOOH were summarized. A suggestion was put forward to construct a “theoretical calculation-machine learning-in situ experiment” system, providing a reference for designing reconstructed catalysts with high stability and product selectivity.关键词:electrocatalytic CO2 reduction;formic acid;p-block metal-based catalysts;in situ reconstruction of catalysts;reconstruction types;in situ characterization techniques94|0|0更新时间:2025-12-25 -
 摘要:Dry reforming of methane and carbon dioxide (DRM) is a promising “carbon-neutral” industrial catalytic process. However, the process suffers from the problem of catalyst deactivation due to coke deposition. Current researches on DRM catalysts primarily focus on single-factor regulation of active metals, while studies on the synergistic regulation between supports and active metals remain relatively limited. Based on the Ni/MgAl2O4 catalyst, a series of Ni-based catalysts and Cs, In doped Ni-based catalysts with the constant Ni mass fraction of 2% were prepared by co-precipitation method. Combining with various characterization techniques such as XRD, H2-TPR, and CO2-TPD, the effects of n(MgO):n(Al2O3) as well as Cs, In doping on catalyst structures and DRM catalytic performances were investigated, and the anti-coking mechanism of catalysts was analyzed. The results indicate that after reaction for 120 h under the conditions of temperature of 1073 K, pressure of 0.1 MPa, V(CO2):V(CH4) of 1:1 and space velocity of 72000 mL/(g·h), the catalyst with n(Cs):n(In):n(Ni) of 1:1:5 and n(MgO):n(Al2O3) of 3:1 (Cs0.2In0.2-Ni/MAN-3) exhibits optimal catalytic and coke resistance performances, while its CH4 conversion rate and CO2 conversion rate are 76.3% and 79.5%, respectively, and the TG weight loss (mass fraction) is 2.0%.关键词:dry reforming reaction;Ni-based catalysts;coke resistance performances;Mg-Al spinel;Cs, In doping67|0|0更新时间:2025-12-25
摘要:Dry reforming of methane and carbon dioxide (DRM) is a promising “carbon-neutral” industrial catalytic process. However, the process suffers from the problem of catalyst deactivation due to coke deposition. Current researches on DRM catalysts primarily focus on single-factor regulation of active metals, while studies on the synergistic regulation between supports and active metals remain relatively limited. Based on the Ni/MgAl2O4 catalyst, a series of Ni-based catalysts and Cs, In doped Ni-based catalysts with the constant Ni mass fraction of 2% were prepared by co-precipitation method. Combining with various characterization techniques such as XRD, H2-TPR, and CO2-TPD, the effects of n(MgO):n(Al2O3) as well as Cs, In doping on catalyst structures and DRM catalytic performances were investigated, and the anti-coking mechanism of catalysts was analyzed. The results indicate that after reaction for 120 h under the conditions of temperature of 1073 K, pressure of 0.1 MPa, V(CO2):V(CH4) of 1:1 and space velocity of 72000 mL/(g·h), the catalyst with n(Cs):n(In):n(Ni) of 1:1:5 and n(MgO):n(Al2O3) of 3:1 (Cs0.2In0.2-Ni/MAN-3) exhibits optimal catalytic and coke resistance performances, while its CH4 conversion rate and CO2 conversion rate are 76.3% and 79.5%, respectively, and the TG weight loss (mass fraction) is 2.0%.关键词:dry reforming reaction;Ni-based catalysts;coke resistance performances;Mg-Al spinel;Cs, In doping67|0|0更新时间:2025-12-25 -
 摘要:As an emerging new energy technology, the direct synthesis of ethanol from syngas still faces problems such as low catalytic efficiency of catalysts and product selectivities. These problems are mainly due to the high energy barriers of C—O bond breakage and C—C bond formation, as well as the difficulty of controlling reaction. So, the reaction mechanisms of direct synthesis of ethanol from syngas on CoCu(100), CoCu(111) and heterojunction CoCu(100)/(111) surfaces were systematically studied by density functional theory (DFT), and the effects of heterojunction CoCu(100)/(111) on CO activation, C—O bond breakage and C—C bond formation were emphatically discussed. The results show that CO is hydrogenated at the Co site to generate CHO, further dissociated and hydrogenated to generate intermediate CHx, and CO is then inserted into intermediate CHx to generate intermediate CHxCO, and finally converted to ethanol by continuous hydrogenation. Compared with CoCu(100) and CoCu(111) surfaces, heterojunction CoCu(100)/(111) surface has advantages in promoting CO activation, C—O bond breakage and C—C bond formation due to its good local electronic structure. The results can provide an important theoretical basis for the design and optimization of high-efficiency Cu-based catalysts, and provide reference for the industrial application of synthesis of ethanol from syngas.关键词:catalytic mechanism;density functional theory;heterojunction CoCu(100)/(111) surface;local electronic structure optimization42|0|0更新时间:2025-12-25
摘要:As an emerging new energy technology, the direct synthesis of ethanol from syngas still faces problems such as low catalytic efficiency of catalysts and product selectivities. These problems are mainly due to the high energy barriers of C—O bond breakage and C—C bond formation, as well as the difficulty of controlling reaction. So, the reaction mechanisms of direct synthesis of ethanol from syngas on CoCu(100), CoCu(111) and heterojunction CoCu(100)/(111) surfaces were systematically studied by density functional theory (DFT), and the effects of heterojunction CoCu(100)/(111) on CO activation, C—O bond breakage and C—C bond formation were emphatically discussed. The results show that CO is hydrogenated at the Co site to generate CHO, further dissociated and hydrogenated to generate intermediate CHx, and CO is then inserted into intermediate CHx to generate intermediate CHxCO, and finally converted to ethanol by continuous hydrogenation. Compared with CoCu(100) and CoCu(111) surfaces, heterojunction CoCu(100)/(111) surface has advantages in promoting CO activation, C—O bond breakage and C—C bond formation due to its good local electronic structure. The results can provide an important theoretical basis for the design and optimization of high-efficiency Cu-based catalysts, and provide reference for the industrial application of synthesis of ethanol from syngas.关键词:catalytic mechanism;density functional theory;heterojunction CoCu(100)/(111) surface;local electronic structure optimization42|0|0更新时间:2025-12-25 -
 摘要:Direct synthesis of α-olefins through CO hydrogenation catalyzed by Fe-based catalysts is one of the important ways to realize carbon resource utilization. xFe/Zn and xFe/Zn-yNa catalysts were prepared by co-precipitation and iso-volume impregnation methods, respectively, and they were evaluated by a sixteen-channel high-throughput reactor to realize rapid and efficient screening of catalysts. The promotional effects of Zn and Na additives on the preparation of α-olefins by Fe-based catalysts were also investigated. The catalysts before and after reaction were characterized by N2 adsorption/desorption, XRD, H2-TPR and so on. The data shows that Zn can form ZnFe2O4 solid solution with Fe during the addition process, improving the stability of catalysts. The addition of Na facilitates the activation of catalysts, promoting the formation of the active phases. 1.0Fe/Zn-3.6Na ((n(Fe)/n(Zn) = 1.0 and w(Na) = 3.6%) has the optimal catalytic performance under the reaction conditions of 340 ℃, 2.0 MPa and volume space velocity of 54000 mL/(g·h): CO conversion rate of 48.17%, CH4 selectivity of 6.28%, alkene to alkane selectivity ratio for C2~C4 hydrocarbon products (O/P value) of 6.25 and mass fraction of α-olefins in C5~C15 hydrocarbon products increasing to 38.19% (data collection time of 48 h). For the screening of sixteen catalysts, high-throughput reactors can save about 94% of experimental time, compared with conventional single fixed-bed reactors, and perform excellently in stable control of reaction temperatures, pressures and other aspects of highly exothermic reactions.关键词:CO hydrogenation;Fe-based catalysts;α-olefins;promoters75|0|0更新时间:2025-12-25
摘要:Direct synthesis of α-olefins through CO hydrogenation catalyzed by Fe-based catalysts is one of the important ways to realize carbon resource utilization. xFe/Zn and xFe/Zn-yNa catalysts were prepared by co-precipitation and iso-volume impregnation methods, respectively, and they were evaluated by a sixteen-channel high-throughput reactor to realize rapid and efficient screening of catalysts. The promotional effects of Zn and Na additives on the preparation of α-olefins by Fe-based catalysts were also investigated. The catalysts before and after reaction were characterized by N2 adsorption/desorption, XRD, H2-TPR and so on. The data shows that Zn can form ZnFe2O4 solid solution with Fe during the addition process, improving the stability of catalysts. The addition of Na facilitates the activation of catalysts, promoting the formation of the active phases. 1.0Fe/Zn-3.6Na ((n(Fe)/n(Zn) = 1.0 and w(Na) = 3.6%) has the optimal catalytic performance under the reaction conditions of 340 ℃, 2.0 MPa and volume space velocity of 54000 mL/(g·h): CO conversion rate of 48.17%, CH4 selectivity of 6.28%, alkene to alkane selectivity ratio for C2~C4 hydrocarbon products (O/P value) of 6.25 and mass fraction of α-olefins in C5~C15 hydrocarbon products increasing to 38.19% (data collection time of 48 h). For the screening of sixteen catalysts, high-throughput reactors can save about 94% of experimental time, compared with conventional single fixed-bed reactors, and perform excellently in stable control of reaction temperatures, pressures and other aspects of highly exothermic reactions.关键词:CO hydrogenation;Fe-based catalysts;α-olefins;promoters75|0|0更新时间:2025-12-25 -
 摘要:Ethylene oligomerization is the primary route for industrial synthesis of higher carbon α-olefins. Traditional homogeneous catalytic systems face challenges such as difficult in catalyst separation and poor product selectivity, necessitating the development of novel heterogeneous catalytic systems for process optimization. A series of Ni catalysts supported on amorphous aluminum silicates were prepared by sol-gel method + impregnation method, and the effects of different m(SiO2):m(Al2O3) on physicochemical properties of the Ni catalysts and their catalytic performances in ethylene oligomerization were investigated. The results show that under the reaction conditions of temperature of 240 °C, pressure of 1.5 MPa and space velocity of 10000 mL/(g·h) for 6 h, the catalyst (Ni5/Si9-Al11) with Ni mass fraction of 5% and m(SiO2):m(Al2O3) of 9:11 exhibits the best catalytic performance. It achieves ethylene conversion rate of 71.36%, with the products dominate by C4~C8 even-numbered olefins. Notably, the C4 olefins selectivity reaches 81.78%. Differences of m(SiO2):m(Al2O3) in the catalysts have no impact on chemical valence state of Ni, which remains in the Ni2+ form.关键词:ethylene oligomerization;amorphous aluminum silicates;Ni catalysts;synergistic interaction73|0|0更新时间:2025-12-25
摘要:Ethylene oligomerization is the primary route for industrial synthesis of higher carbon α-olefins. Traditional homogeneous catalytic systems face challenges such as difficult in catalyst separation and poor product selectivity, necessitating the development of novel heterogeneous catalytic systems for process optimization. A series of Ni catalysts supported on amorphous aluminum silicates were prepared by sol-gel method + impregnation method, and the effects of different m(SiO2):m(Al2O3) on physicochemical properties of the Ni catalysts and their catalytic performances in ethylene oligomerization were investigated. The results show that under the reaction conditions of temperature of 240 °C, pressure of 1.5 MPa and space velocity of 10000 mL/(g·h) for 6 h, the catalyst (Ni5/Si9-Al11) with Ni mass fraction of 5% and m(SiO2):m(Al2O3) of 9:11 exhibits the best catalytic performance. It achieves ethylene conversion rate of 71.36%, with the products dominate by C4~C8 even-numbered olefins. Notably, the C4 olefins selectivity reaches 81.78%. Differences of m(SiO2):m(Al2O3) in the catalysts have no impact on chemical valence state of Ni, which remains in the Ni2+ form.关键词:ethylene oligomerization;amorphous aluminum silicates;Ni catalysts;synergistic interaction73|0|0更新时间:2025-12-25 -
 摘要:As an important industrial raw material, γ-butyrolactone (GBL) is in increasing demand day by day. Currently, the dehydrogenation of 1,4-butanediol (BDO) on Cu-based catalysts represents the predominant method for GBL preparation. However, the dehydrogenation reaction mechanism on Cu-based catalysts and the influence of the Cu valence states on the reaction remain to be thoroughly investigated. The reaction process of BDO dehydrogenation to GBL on Cu-based catalysts was calculated and analyzed by density functional theory. The energy barriers of different reaction pathways were compared, and the adsorption energies, transition states, etc. of substances on Cu+-perfect type catalyst, Cu+-defect type catalyst, Cu0 catalyst and Cu0/Cu+ composite type catalyst were studied. The results show that the most probable reaction pathway (pathway A) involves BDO first eliminating two hydrogen atoms follows by cyclization to form 2-hydroxytetrahydrofuran (2-HTHF), which then loses two additional hydrogen atoms to produce GBL, with the energy barrier of rate-determining step of pathway A of 287.67 kJ/mol. Under pathway A, Cu+ demonstrates strong adsorption towards BDO and 2-HTHF molecules, while Cu0 exhibits greater aptitude for H2 activation. Both Cu+ and oxygen vacancy effectively facilitate the breaking of O—H and C—H bonds, activating BDO and 2-HTHF, and Cu⁰ significantly promotes H2 synthesis under low H2 coverage conditions. Notably, Cu⁰/Cu+ composite catalyst outperforms the other three catalysts in terms of catalytic performance.关键词:1,4-butanediol;γ-butyrolactone;density functional theory;Cu valence states;oxygen vacancy139|0|0更新时间:2025-12-25
摘要:As an important industrial raw material, γ-butyrolactone (GBL) is in increasing demand day by day. Currently, the dehydrogenation of 1,4-butanediol (BDO) on Cu-based catalysts represents the predominant method for GBL preparation. However, the dehydrogenation reaction mechanism on Cu-based catalysts and the influence of the Cu valence states on the reaction remain to be thoroughly investigated. The reaction process of BDO dehydrogenation to GBL on Cu-based catalysts was calculated and analyzed by density functional theory. The energy barriers of different reaction pathways were compared, and the adsorption energies, transition states, etc. of substances on Cu+-perfect type catalyst, Cu+-defect type catalyst, Cu0 catalyst and Cu0/Cu+ composite type catalyst were studied. The results show that the most probable reaction pathway (pathway A) involves BDO first eliminating two hydrogen atoms follows by cyclization to form 2-hydroxytetrahydrofuran (2-HTHF), which then loses two additional hydrogen atoms to produce GBL, with the energy barrier of rate-determining step of pathway A of 287.67 kJ/mol. Under pathway A, Cu+ demonstrates strong adsorption towards BDO and 2-HTHF molecules, while Cu0 exhibits greater aptitude for H2 activation. Both Cu+ and oxygen vacancy effectively facilitate the breaking of O—H and C—H bonds, activating BDO and 2-HTHF, and Cu⁰ significantly promotes H2 synthesis under low H2 coverage conditions. Notably, Cu⁰/Cu+ composite catalyst outperforms the other three catalysts in terms of catalytic performance.关键词:1,4-butanediol;γ-butyrolactone;density functional theory;Cu valence states;oxygen vacancy139|0|0更新时间:2025-12-25 -
 摘要:Traditional methods for cyclohexanol production, such as phenol hydrogenation method and cyclohexane oxidation method, suffer from high energy consumptions and safety concerns. In contrast, the cyclohexene hydration method has attracted significant attention due to its high cyclohexanol selectivity and low hydrogen consumption. However, existing catalysts (such as ZSM-5 zeolite) for cyclohexene hydration to cyclohexanol face challenges including structural framework instabilities and uneven acid distributions during acid content regulation. A series of catalysts were prepared by compositing ZSM-5 zeolite with dry gel powder (primarily pseudo-boehmite) via extrusion molding. The crystal and textural structures of catalysts were characterized by XRD and N2 physisorption-desorption, etc., and their catalytic performances were evaluated in a fixed-bed reactor. The results show that under optimized conditions of temperature of 110 °C, pressure of 0.1 MPa, N2 flow rate of 10 mL/min, space velocity of 0.33 h-1 and n(water):n(cyclohexene) of 6:1 for 1 h, catalyst Z4-4:1 (m(ZSM-5):m(dry gel powder) of 4:1) exhibits superior catalytic performance with cyclohexene conversion rate of 8.2% and cyclohexanol selectivity of 79.0%. The catalytic performance of catalyst can be improved by enhancing acid strength and increasing acid quantity within specific ranges.关键词:zeolites;cyclohexene;cyclohexanol;acid contents134|0|0更新时间:2025-12-25
摘要:Traditional methods for cyclohexanol production, such as phenol hydrogenation method and cyclohexane oxidation method, suffer from high energy consumptions and safety concerns. In contrast, the cyclohexene hydration method has attracted significant attention due to its high cyclohexanol selectivity and low hydrogen consumption. However, existing catalysts (such as ZSM-5 zeolite) for cyclohexene hydration to cyclohexanol face challenges including structural framework instabilities and uneven acid distributions during acid content regulation. A series of catalysts were prepared by compositing ZSM-5 zeolite with dry gel powder (primarily pseudo-boehmite) via extrusion molding. The crystal and textural structures of catalysts were characterized by XRD and N2 physisorption-desorption, etc., and their catalytic performances were evaluated in a fixed-bed reactor. The results show that under optimized conditions of temperature of 110 °C, pressure of 0.1 MPa, N2 flow rate of 10 mL/min, space velocity of 0.33 h-1 and n(water):n(cyclohexene) of 6:1 for 1 h, catalyst Z4-4:1 (m(ZSM-5):m(dry gel powder) of 4:1) exhibits superior catalytic performance with cyclohexene conversion rate of 8.2% and cyclohexanol selectivity of 79.0%. The catalytic performance of catalyst can be improved by enhancing acid strength and increasing acid quantity within specific ranges.关键词:zeolites;cyclohexene;cyclohexanol;acid contents134|0|0更新时间:2025-12-25 -
 摘要:Cu/SiO2 exhibits excellent catalytic performance and is widely used in dehydrogenation of cyclohexanol to cyclohexanone. However, Cu/SiO2 tends to sinter at high temperatures, leading to agglomeration of the active components and a shortened service life. Cu/SiO2 with different Cu loadings (calculated by mass fraction of CuO) were prepared using the co-precipitation method. Based on Cu/SiO2 with the Cu loading of 10.0%, Cu-Zn/SiO2, Cu-Al/SiO2 and Cu-Zn-Al/SiO2 were further prepared. The catalytic performances of catalysts in dehydrogenation of cyclohexanol to cyclohexanone were studied. And the crystal structures and textural properties of catalysts were characterized by XRD and N2 adsorption/desorption, etc. The results show that Cu-Zn-Al/SiO2 (m(CuO):m(ZnO):m(Al2O3) = 10:2:1) exhibits the best catalytic performance with cyclohexanol conversion rate, cyclohexanone selectivity and cyclohexanone yield of 72.3%, 98.3% and 71.1%, respectively, under the reaction conditions of reaction temperature of 240 °C, reaction pressure of 1.01 × 105 Pa and space velocity of 3.6 h-1 for 4 h, Additionally, the catalyst maintains stable catalytic performance over 24 h. The introduction of ZnO in Cu/SiO2 can inhibit the migration and aggregation of the active component Cu, improves its dispersion during the reduction process and slows down Cu grain growth. The introduction of Al2O3 can enhance the interaction between the active component Cu and the SiO2 support, thereby improving thermal stability of the catalyst.关键词:Cu-based catalysts;dehydrogenation of cyclohexanol;cyclohexanone;thermal stability39|0|0更新时间:2025-12-25
摘要:Cu/SiO2 exhibits excellent catalytic performance and is widely used in dehydrogenation of cyclohexanol to cyclohexanone. However, Cu/SiO2 tends to sinter at high temperatures, leading to agglomeration of the active components and a shortened service life. Cu/SiO2 with different Cu loadings (calculated by mass fraction of CuO) were prepared using the co-precipitation method. Based on Cu/SiO2 with the Cu loading of 10.0%, Cu-Zn/SiO2, Cu-Al/SiO2 and Cu-Zn-Al/SiO2 were further prepared. The catalytic performances of catalysts in dehydrogenation of cyclohexanol to cyclohexanone were studied. And the crystal structures and textural properties of catalysts were characterized by XRD and N2 adsorption/desorption, etc. The results show that Cu-Zn-Al/SiO2 (m(CuO):m(ZnO):m(Al2O3) = 10:2:1) exhibits the best catalytic performance with cyclohexanol conversion rate, cyclohexanone selectivity and cyclohexanone yield of 72.3%, 98.3% and 71.1%, respectively, under the reaction conditions of reaction temperature of 240 °C, reaction pressure of 1.01 × 105 Pa and space velocity of 3.6 h-1 for 4 h, Additionally, the catalyst maintains stable catalytic performance over 24 h. The introduction of ZnO in Cu/SiO2 can inhibit the migration and aggregation of the active component Cu, improves its dispersion during the reduction process and slows down Cu grain growth. The introduction of Al2O3 can enhance the interaction between the active component Cu and the SiO2 support, thereby improving thermal stability of the catalyst.关键词:Cu-based catalysts;dehydrogenation of cyclohexanol;cyclohexanone;thermal stability39|0|0更新时间:2025-12-25 -
 摘要:In response to the increasingly urgent carbon reduction demand of the non-ferrous metal smelting industry, the latest research progress in carbon reduction technologies across the entire smelting process was reviewed systematically, including key stages such as pre-smelting, during smelting and post-smelting. Through a comparative analysis of mainstream technologies such as absorption, solid adsorption and membrane separation methods, it identifies the key factors restricting the industrial application of carbon dioxide (CO2) separation technologies, mainly including separation cost, process complexity and product purity. In this regard, relevant studies have innovatively applied liquid membrane coupling technology and pressure swing adsorption technology, aiming to improve the separation efficiency of CO2 from non-ferrous metal smelting flue gases. These technologies can provide new solutions to overcome the technical bottlenecks of CO2 resource utilization and are expected to contribute to the realization of the “carbon peaking and carbon neutrality” goals.关键词:CO2;non-ferrous metal smelting;separation and purification;carbon reduction80|0|0更新时间:2025-12-25
摘要:In response to the increasingly urgent carbon reduction demand of the non-ferrous metal smelting industry, the latest research progress in carbon reduction technologies across the entire smelting process was reviewed systematically, including key stages such as pre-smelting, during smelting and post-smelting. Through a comparative analysis of mainstream technologies such as absorption, solid adsorption and membrane separation methods, it identifies the key factors restricting the industrial application of carbon dioxide (CO2) separation technologies, mainly including separation cost, process complexity and product purity. In this regard, relevant studies have innovatively applied liquid membrane coupling technology and pressure swing adsorption technology, aiming to improve the separation efficiency of CO2 from non-ferrous metal smelting flue gases. These technologies can provide new solutions to overcome the technical bottlenecks of CO2 resource utilization and are expected to contribute to the realization of the “carbon peaking and carbon neutrality” goals.关键词:CO2;non-ferrous metal smelting;separation and purification;carbon reduction80|0|0更新时间:2025-12-25 -
 摘要:Tail gas from Fe-Mo methanol-to-formaldehyde production contains multiple pollutants such as carbon monoxide (CO), formaldehyde (CH2O), methanol (CH3OH) and dimethyl ether (DME), which are difficult to remove collaboratively. Based on the characteristics of different industrial catalytic combustion catalysts, a combination loading strategy was adopted to enhance the synergistic purification efficiency of complex pollutants. The physical properties of two industrial catalysts, Cat 1# and Cat 2#, were analyzed using XRD, N2 adsorption/desorption and TEM. Combined with laboratory activity evaluation results and the operation of the industrial emission control system (ECS), the structure-activity relationships of the catalysts were clarified, and their industrial application performance was elucidated. The results show that Cat 1# has a larger specific surface area, which promotes precious metal dispersion and reduces the light-off temperature of CO, and Cat 2# has a larger average pore size, which decreases the diffusion resistance of DME and improves its conversion rate. The combination loading strategy integrates the advantages of both catalysts. Laboratory evaluation results show that the reaction start to light off at a set temperature of 150 ℃ (bed temperature 135.4 ℃). After light-off, the heat released by CO catalytic combustion increases the bed temperature from 100 ℃ to 110 ℃, and the DME conversion rate exceeds 90%. In industrial operation, at the initial inlet temperature of 170 ℃, the outlet formaldehyde mass concentration is 1.1 mg/m3 and the total non-methane hydrocarbons mass concentration is 1.7 mg/m3. At the end of operation, when the inlet temperature is increased to 183 ℃, the outlet formaldehyde mass concentration reaches 4.2 mg/m3 and the total non-methane hydrocarbons mass concentration is 9.8 mg/m3, still meeting the emission standards. The combination loading strategy provides a new approach for catalytic purification of complex waste gas, and its industrial application data offers important reference value for engineering scale-up.关键词:formaldehyde production tail gas;precious metal catalyst;multiple pollutants;catalytic combustion29|0|0更新时间:2025-12-25
摘要:Tail gas from Fe-Mo methanol-to-formaldehyde production contains multiple pollutants such as carbon monoxide (CO), formaldehyde (CH2O), methanol (CH3OH) and dimethyl ether (DME), which are difficult to remove collaboratively. Based on the characteristics of different industrial catalytic combustion catalysts, a combination loading strategy was adopted to enhance the synergistic purification efficiency of complex pollutants. The physical properties of two industrial catalysts, Cat 1# and Cat 2#, were analyzed using XRD, N2 adsorption/desorption and TEM. Combined with laboratory activity evaluation results and the operation of the industrial emission control system (ECS), the structure-activity relationships of the catalysts were clarified, and their industrial application performance was elucidated. The results show that Cat 1# has a larger specific surface area, which promotes precious metal dispersion and reduces the light-off temperature of CO, and Cat 2# has a larger average pore size, which decreases the diffusion resistance of DME and improves its conversion rate. The combination loading strategy integrates the advantages of both catalysts. Laboratory evaluation results show that the reaction start to light off at a set temperature of 150 ℃ (bed temperature 135.4 ℃). After light-off, the heat released by CO catalytic combustion increases the bed temperature from 100 ℃ to 110 ℃, and the DME conversion rate exceeds 90%. In industrial operation, at the initial inlet temperature of 170 ℃, the outlet formaldehyde mass concentration is 1.1 mg/m3 and the total non-methane hydrocarbons mass concentration is 1.7 mg/m3. At the end of operation, when the inlet temperature is increased to 183 ℃, the outlet formaldehyde mass concentration reaches 4.2 mg/m3 and the total non-methane hydrocarbons mass concentration is 9.8 mg/m3, still meeting the emission standards. The combination loading strategy provides a new approach for catalytic purification of complex waste gas, and its industrial application data offers important reference value for engineering scale-up.关键词:formaldehyde production tail gas;precious metal catalyst;multiple pollutants;catalytic combustion29|0|0更新时间:2025-12-25 -
 摘要:Liquid amine absorption is one of the main technologies currently used in industry to capture CO2 from flue gas, but it suffers from high energy consumption, volatility and secondary pollution. Porous liquids (PLs), which combine the characteristics of porous solids and flowing liquids, can reduce the energy consumption of CO2 capture and suppress adsorbent volatilization. However, their high viscosity limits practical application. Poly(1-vinyl-3-butylimidazolium chloride) (P[VBIm]Cl) was combined with polyethylene glycol-200 (PEG-200), and a metal-organic framework material ZIF-8 was introduced to successfully construct a series of low-viscosity porous deep eutectic solvents (PDES), referred to as ZIF-8-PDES-X% (X% represents the mass fraction of ZIF-8). Among them, the optimized ZIF-8-PDES-5% exhibits a CO2 adsorption capacity of 0.42 mmol/g under ambient temperature and pressure, representing a 16.7% increase compared to pure deep eutectic solvent (DES). Moreover, over 98% desorption efficiency can be achieved at room temperature by purging with Ar gas. After five adsorption-desorption cycles, the CO2 adsorption rate of ZIF-8-PDES-5% can remain above 84%. Meanwhile, the viscosity of this system is only 2760 mPa·s. This study effectively addresses the high-viscosity limitation of porous liquids and provides a new research strategy for environmentally friendly and low-cost CO2 capture processes.关键词:ZIF-8;PEG-200;CO2 capture;deep eutectic solvent;low viscosity;high desorption rate63|0|0更新时间:2025-12-25
摘要:Liquid amine absorption is one of the main technologies currently used in industry to capture CO2 from flue gas, but it suffers from high energy consumption, volatility and secondary pollution. Porous liquids (PLs), which combine the characteristics of porous solids and flowing liquids, can reduce the energy consumption of CO2 capture and suppress adsorbent volatilization. However, their high viscosity limits practical application. Poly(1-vinyl-3-butylimidazolium chloride) (P[VBIm]Cl) was combined with polyethylene glycol-200 (PEG-200), and a metal-organic framework material ZIF-8 was introduced to successfully construct a series of low-viscosity porous deep eutectic solvents (PDES), referred to as ZIF-8-PDES-X% (X% represents the mass fraction of ZIF-8). Among them, the optimized ZIF-8-PDES-5% exhibits a CO2 adsorption capacity of 0.42 mmol/g under ambient temperature and pressure, representing a 16.7% increase compared to pure deep eutectic solvent (DES). Moreover, over 98% desorption efficiency can be achieved at room temperature by purging with Ar gas. After five adsorption-desorption cycles, the CO2 adsorption rate of ZIF-8-PDES-5% can remain above 84%. Meanwhile, the viscosity of this system is only 2760 mPa·s. This study effectively addresses the high-viscosity limitation of porous liquids and provides a new research strategy for environmentally friendly and low-cost CO2 capture processes.关键词:ZIF-8;PEG-200;CO2 capture;deep eutectic solvent;low viscosity;high desorption rate63|0|0更新时间:2025-12-25 -
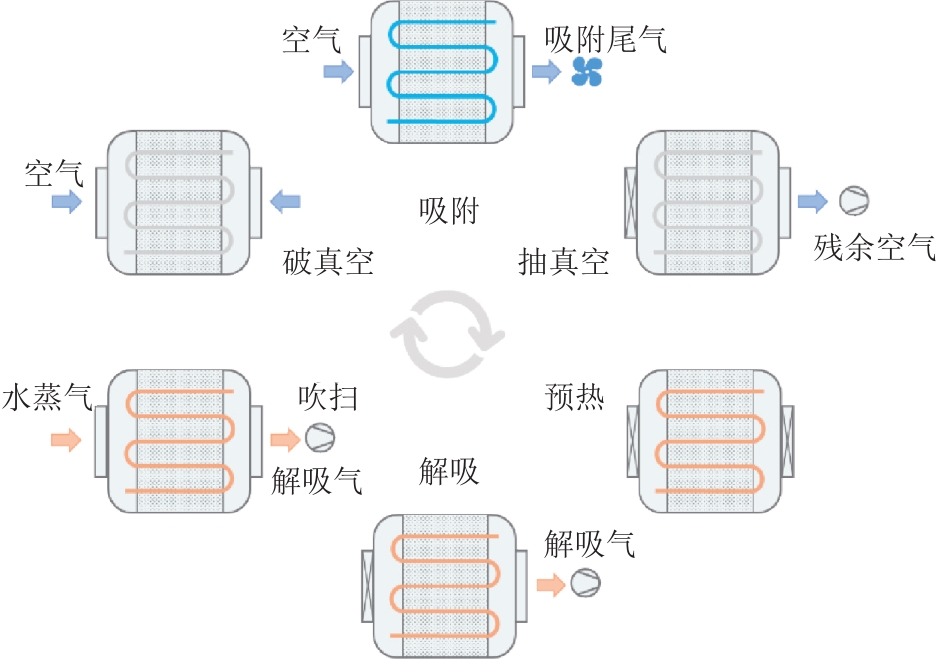 摘要:Steam-assisted temperature vacuum swing adsorption (sTVSA) cycle for CO2 capture is a promising direct air capture (DAC) technology, featuring fast desorption kinetics, high driving potential difference and inhibition of water desorption. To explore the application potential of the sTVSA cycle in DAC systems, a six-step sTVSA cycle (referred to as the “sTVSA cycle”) was proposed. Based on the constructed adsorption bed and adsorption/desorption model, thermodynamic, kinetic and energy consumption analyses were conducted. Simulation results show that, compared with the traditional temperature vacuum swing adsorption (TVSA) cycle, the sTVSA cycle can nearly double the system’s CO2 cyclic capacity under the same desorption temperature and pressure, accelerate desorption kinetics, and significantly improve system’s CO2 production. In addition, steam purging reduces the system requirements for desorption temperature and pressure, lowering the required desorption temperature from 100 ℃ to 90 ℃ and increasing the desorption pressure from 3 kPa to 5 kPa. Compared to a simple TVSA cycle, energy consumption is reduced by approximately 19%. Therefore, constructing a DAC system based on the sTVSA cycle can effectively reduce equipment investment and operational costs, promote the engineering application of DAC systems, and demonstrates promising technical prospects.关键词:steam-assisted temperature vacuum swing adsorption;direct air capture;thermodynamics;kinetics;energy consumption;simulation136|0|0更新时间:2025-12-25
摘要:Steam-assisted temperature vacuum swing adsorption (sTVSA) cycle for CO2 capture is a promising direct air capture (DAC) technology, featuring fast desorption kinetics, high driving potential difference and inhibition of water desorption. To explore the application potential of the sTVSA cycle in DAC systems, a six-step sTVSA cycle (referred to as the “sTVSA cycle”) was proposed. Based on the constructed adsorption bed and adsorption/desorption model, thermodynamic, kinetic and energy consumption analyses were conducted. Simulation results show that, compared with the traditional temperature vacuum swing adsorption (TVSA) cycle, the sTVSA cycle can nearly double the system’s CO2 cyclic capacity under the same desorption temperature and pressure, accelerate desorption kinetics, and significantly improve system’s CO2 production. In addition, steam purging reduces the system requirements for desorption temperature and pressure, lowering the required desorption temperature from 100 ℃ to 90 ℃ and increasing the desorption pressure from 3 kPa to 5 kPa. Compared to a simple TVSA cycle, energy consumption is reduced by approximately 19%. Therefore, constructing a DAC system based on the sTVSA cycle can effectively reduce equipment investment and operational costs, promote the engineering application of DAC systems, and demonstrates promising technical prospects.关键词:steam-assisted temperature vacuum swing adsorption;direct air capture;thermodynamics;kinetics;energy consumption;simulation136|0|0更新时间:2025-12-25 -
 摘要:The phase change absorption process is an effective way to reduce the energy consumption of CO2 capture by organic amines. Based on piperazine (PZ)-n-butanol liquid-solid phase change CO2 absorbent, 2-amino-2-methyl-1-propanol (AMP) was used to improve its desorption performance, and the PZ-AMP-n-butanol ternary phase change absorbent was constrcted. The absorption rate, absorption quantity, desorption rate, and desorption amount were used as indicators to optimize the molar concentration ratio of PZ to AMP. The influence of absorption and desorption temperature was analyzed, and the absorbent cycle stability was evaluated, and the phase change mechanism of the ternary absorption system was analyzed. The results show that the PZ-AMP-n-butanol absorbent has good phase separation, absorption and desorption performances. When the molar concentration ratio of PZ to AMP is 3.0:0.5, the CO2 absorption amount and absorption load of PZ-AMP-n-butanol reach their maximum values, which are 2.92 mol/L and 0.84 mol/mol, respectively, with 44.2% and 51.1% higher than those of 30% (mass fraction) MEA aqueous solution. Under the action of AMP (the molar concentration ratio of PZ to AMP is 3.0:0.5), the desorption rate at 120 ℃ is 73.2%, which is 18.8% higher than that of PZ-n-butanol binary absorbent. The load capacity after five absorption-desorption cycles is 1.84 mol/L, which is 63.3% higher than that of 30% MEA. PZ and AMP absorb CO2 to form the corresponding carbamate and separates out to form a phase change.关键词:CO2 capture;liquid-solid phase transition;PZ-AMP-n-butanol43|0|0更新时间:2025-12-25
摘要:The phase change absorption process is an effective way to reduce the energy consumption of CO2 capture by organic amines. Based on piperazine (PZ)-n-butanol liquid-solid phase change CO2 absorbent, 2-amino-2-methyl-1-propanol (AMP) was used to improve its desorption performance, and the PZ-AMP-n-butanol ternary phase change absorbent was constrcted. The absorption rate, absorption quantity, desorption rate, and desorption amount were used as indicators to optimize the molar concentration ratio of PZ to AMP. The influence of absorption and desorption temperature was analyzed, and the absorbent cycle stability was evaluated, and the phase change mechanism of the ternary absorption system was analyzed. The results show that the PZ-AMP-n-butanol absorbent has good phase separation, absorption and desorption performances. When the molar concentration ratio of PZ to AMP is 3.0:0.5, the CO2 absorption amount and absorption load of PZ-AMP-n-butanol reach their maximum values, which are 2.92 mol/L and 0.84 mol/mol, respectively, with 44.2% and 51.1% higher than those of 30% (mass fraction) MEA aqueous solution. Under the action of AMP (the molar concentration ratio of PZ to AMP is 3.0:0.5), the desorption rate at 120 ℃ is 73.2%, which is 18.8% higher than that of PZ-n-butanol binary absorbent. The load capacity after five absorption-desorption cycles is 1.84 mol/L, which is 63.3% higher than that of 30% MEA. PZ and AMP absorb CO2 to form the corresponding carbamate and separates out to form a phase change.关键词:CO2 capture;liquid-solid phase transition;PZ-AMP-n-butanol43|0|0更新时间:2025-12-25 -
 摘要:Shipboard carbon capture, utilization, and storage (CCUS) technology represents an effective solution for large-scale decarbonization in the shipping industry. However, its practical application faces significant challenges due to spatial constraint, regeneration energy consumption, and cost issue. Developing amine solution absorption systems with superior CO2 absorption capacity, cycling stability, and low regeneration energy consumption is a critical pathway to address these application barriers. Targeting the characteristics of low CO2 partial pressure and high volume in ship exhaust, a series of single component, binary compound and ternary compound amine solution systems were prepared using N-aminoethyl piperazine (AEP), hydroxyethyl ethylenediamine (AEEA) and N-methyldiethanolamine (MDEA) as amine components, and their CO2 absorption and regeneration performances were investigated. The results show that the ternary compound amine solution system (formulation I) with mass fraction of AEP, AEEA, MDEA and H2O of 35%, 16%, 9% and 40%, respectively, exhibits optimal CO2 absorption and regeneration performance. At the absorption temperature of 40 °C, CO2 mole absorption capacity, volumetric absorption capacity and absorption rate of formulation I are 1.023 mol/mol, 114.5 mL/g and 2.13 × 10-5 mol/(g·min), respectively, representing increases of 84.3%, 87.4% and 1.75% compared to monoethanolamine (MEA) solution (w = 30%). At the regeneration temperature of 105 ℃, formulation I achieves the maximum regeneration rate, CO2 regeneration capacity and regeneration efficiency of 4.68 × 10-5 mol/(g·min), 0.919 mol/mol and 89.9%, respectively, corresponding to improvements of 35.3%, 64.3% and 15.6% over MEA solution. Furthermore, formulation I maintains good cycling stability, retaining approximately regeneration efficiency of 90% after 10 absorption-regeneration cycles. The regeneration energy consumption of formulation I in the first absorption-regeneration is 2.54 GJ/t, which is 34.7% lower than that of MEA solution.关键词:compound amine solution systems;CO2 capture;absorption-regeneration performances;ship exhaust102|0|0更新时间:2025-12-25
摘要:Shipboard carbon capture, utilization, and storage (CCUS) technology represents an effective solution for large-scale decarbonization in the shipping industry. However, its practical application faces significant challenges due to spatial constraint, regeneration energy consumption, and cost issue. Developing amine solution absorption systems with superior CO2 absorption capacity, cycling stability, and low regeneration energy consumption is a critical pathway to address these application barriers. Targeting the characteristics of low CO2 partial pressure and high volume in ship exhaust, a series of single component, binary compound and ternary compound amine solution systems were prepared using N-aminoethyl piperazine (AEP), hydroxyethyl ethylenediamine (AEEA) and N-methyldiethanolamine (MDEA) as amine components, and their CO2 absorption and regeneration performances were investigated. The results show that the ternary compound amine solution system (formulation I) with mass fraction of AEP, AEEA, MDEA and H2O of 35%, 16%, 9% and 40%, respectively, exhibits optimal CO2 absorption and regeneration performance. At the absorption temperature of 40 °C, CO2 mole absorption capacity, volumetric absorption capacity and absorption rate of formulation I are 1.023 mol/mol, 114.5 mL/g and 2.13 × 10-5 mol/(g·min), respectively, representing increases of 84.3%, 87.4% and 1.75% compared to monoethanolamine (MEA) solution (w = 30%). At the regeneration temperature of 105 ℃, formulation I achieves the maximum regeneration rate, CO2 regeneration capacity and regeneration efficiency of 4.68 × 10-5 mol/(g·min), 0.919 mol/mol and 89.9%, respectively, corresponding to improvements of 35.3%, 64.3% and 15.6% over MEA solution. Furthermore, formulation I maintains good cycling stability, retaining approximately regeneration efficiency of 90% after 10 absorption-regeneration cycles. The regeneration energy consumption of formulation I in the first absorption-regeneration is 2.54 GJ/t, which is 34.7% lower than that of MEA solution.关键词:compound amine solution systems;CO2 capture;absorption-regeneration performances;ship exhaust102|0|0更新时间:2025-12-25 -
 摘要:Methylcyclohexane(MCH)-toluene liquid hydrogen storage is one of the safe and efficient low-cost hydrogen storage and transportation technologies. MCH dehydrogenation catalyst is the bottleneck of its industrial application. Exploring main reason for deactivation of Pt/Al2O3 catalyst for MCH dehydrogenation can provide reference for the development of high-performance catalysts. Using the formed Pt/γ-Al2O3 pellets as the model catalyst, the catalytic performance of the catalyst for 190 h was studied. The physical and chemical properties of the catalyst before and after the reaction were characterized, and the reasons for its deactivation were analyzed. The results show that the MCH conversion rate decreases from 97.56% to 76.70% after 190 h reaction, and the conversion recoveres to 90.00% after carbon removal regeneration, which confirms that carbon deposition is one of the main reasons for catalyst deactivation. It is found that the crystallite size of Pt particles grows up and the dispersion of Pt particles decreases from 55.36% to 34.09%, which is also the main cause of catalyst deactivation. In the future, it can be considered to optimize the design of dehydrogenation catalyst through the modification of carrier and doping of additives.关键词:methylcyclohexane;dehydrogenation reaction;Pt/Al2O3;catalyst deactivation;carbon deposition46|0|0更新时间:2025-12-25
摘要:Methylcyclohexane(MCH)-toluene liquid hydrogen storage is one of the safe and efficient low-cost hydrogen storage and transportation technologies. MCH dehydrogenation catalyst is the bottleneck of its industrial application. Exploring main reason for deactivation of Pt/Al2O3 catalyst for MCH dehydrogenation can provide reference for the development of high-performance catalysts. Using the formed Pt/γ-Al2O3 pellets as the model catalyst, the catalytic performance of the catalyst for 190 h was studied. The physical and chemical properties of the catalyst before and after the reaction were characterized, and the reasons for its deactivation were analyzed. The results show that the MCH conversion rate decreases from 97.56% to 76.70% after 190 h reaction, and the conversion recoveres to 90.00% after carbon removal regeneration, which confirms that carbon deposition is one of the main reasons for catalyst deactivation. It is found that the crystallite size of Pt particles grows up and the dispersion of Pt particles decreases from 55.36% to 34.09%, which is also the main cause of catalyst deactivation. In the future, it can be considered to optimize the design of dehydrogenation catalyst through the modification of carrier and doping of additives.关键词:methylcyclohexane;dehydrogenation reaction;Pt/Al2O3;catalyst deactivation;carbon deposition46|0|0更新时间:2025-12-25 -
 摘要:Alkaline water electrolysis (ALK) for green hydrogen production is one of the highly promising alternative to fossil fuels. To further advance its industrialization, research has focused on developing non-noble metal catalysts for both cathode and anode, as these materials are cost-effective. Among non-noble metal catalysts, transition metal hydroxide catalysts are particularly well-suited for alkaline water electrolysis due to their simple preparation process and better electrocatalytic performance. Herein, Fe, Co, Ni, Zn, and Mo metal salts were used as raw materials to obtain FeCoNiZnMo quinary hydroxides with a nanoflower structure via pulsed electrodeposition on a Ni mesh. The physical and chemical characterizations of FeCoNiZnMo quinary hydroxides were analyzed by SEM, TEM and XPS. Compared to the low-entropy quaternary hydroxides, the coupling effect among quinary metals in the high-entropy hydroxides could simultaneously increase the oxidation states of nickel and cobalt elements, which is conducive to enhancing its activity of oxygen evolution and hydrogen evolution. Additionally, the oxygen evolution reaction and hydrogen evolution reaction performances of the FeCoNiZnMo hydroxides in 1 mol/L NaOH solution were assessed, alongside a two-electrode stability test for overall water splitting. The results indicate that FeCoNiZnMo hydroxides exhibit oxygen evolution reaction and hydrogen reaction overpotentials of 307 mV and 223 mV at current density of 100 mA/cm2, respectively. Furthermore, FeCoNiZnMo hydroxides exhibit long-term stability for 200 h at 200 mA/cm2.关键词:water electrolysis;oxygen evolution reaction;hydrogen evolution reaction;transition metal hydroxide;bifunctional electrocatalyst;high-entropy46|0|0更新时间:2025-12-25
摘要:Alkaline water electrolysis (ALK) for green hydrogen production is one of the highly promising alternative to fossil fuels. To further advance its industrialization, research has focused on developing non-noble metal catalysts for both cathode and anode, as these materials are cost-effective. Among non-noble metal catalysts, transition metal hydroxide catalysts are particularly well-suited for alkaline water electrolysis due to their simple preparation process and better electrocatalytic performance. Herein, Fe, Co, Ni, Zn, and Mo metal salts were used as raw materials to obtain FeCoNiZnMo quinary hydroxides with a nanoflower structure via pulsed electrodeposition on a Ni mesh. The physical and chemical characterizations of FeCoNiZnMo quinary hydroxides were analyzed by SEM, TEM and XPS. Compared to the low-entropy quaternary hydroxides, the coupling effect among quinary metals in the high-entropy hydroxides could simultaneously increase the oxidation states of nickel and cobalt elements, which is conducive to enhancing its activity of oxygen evolution and hydrogen evolution. Additionally, the oxygen evolution reaction and hydrogen evolution reaction performances of the FeCoNiZnMo hydroxides in 1 mol/L NaOH solution were assessed, alongside a two-electrode stability test for overall water splitting. The results indicate that FeCoNiZnMo hydroxides exhibit oxygen evolution reaction and hydrogen reaction overpotentials of 307 mV and 223 mV at current density of 100 mA/cm2, respectively. Furthermore, FeCoNiZnMo hydroxides exhibit long-term stability for 200 h at 200 mA/cm2.关键词:water electrolysis;oxygen evolution reaction;hydrogen evolution reaction;transition metal hydroxide;bifunctional electrocatalyst;high-entropy46|0|0更新时间:2025-12-25

- Postal code:610225
- Tel:028-85964717 Email:236764603@qq.com
- Technical support is provided by Beijing Founder electronics co., LTD 蜀ICP备12008600号-10
 蜀公网安备51012202001533
蜀公网安备51012202001533 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
- Total Visits:348184 Visits Today:346

0